Quantum mechanical study on the kinetics and thermodynamics of O-alkyl S-methyl dithiocarbonates through α- and β-elimination pathways
Oluwaseun Timothy ESAN1, Isaiah Ajibade ADEJORO1,2, Babatunde Temitope OGUNYEMI1,3*, Oluwatoba Emmanuel OYENEYIN1,4
1Chemistry Department, Faculty of Science, University of Ibadan, Ibadan, Oyo State, Nigeria.
2Department of Chemistry, University of Medical Sciences, Laje Road, Ondo City, Ondo State, Nigeria.
3Chemistry Department, Federal University Otuoke, Bayelsa State, Nigeria.
4Department of Chemical Sciences, Adekunle Ajasin University, Akungba-Akoko, Ondo State, Nigeria.
E-mails: Timothy.esan@yahoo.com (OTE), ajibadejoro@yahoo.com (IAA), btogunyemi@yahoo.com (*BTO), emmanueltoba90@gmail.com (OEO)
*Corresponding Author phone: +2348035811574
Abstract
The study of α- and β-elimination pathways through pyrolysis on the kinetics and thermodynamics of O-alkyl S-methyl Xanthates in which the alkyl groups are ethyl, n-propyl, i-propyl, n-butyl, i-butyl and t-butyl were carried out in the gas-phase using density functional theory (DFT) with B3LYP at 6-311++G** using Spartan. The mechanism proceeded through a six-centered cyclic transition state. The result obtained for the C-H, C-O and S-H bond lengthening character in a single step suggested that the reaction is a concerted and asynchronous. The energy of formation for the αelimination pathways is lower compared to the β-elimination initiated from the hydrogen of the β-carbon. The activation parameters obtained for the β-elimination pathways ranged from ∆H* (161.34-133.87 kJ/mol), ∆S* (-31.18-25.65 J/mol/K), ∆G* (180.65-149.77 kJ/mol) Ea (166.49-136.82 kJ/mol), k =1.14·10-3-6.0·10-2S-2), A (3.06·1011-5.89·1011). In addition, α-elimination pathway activation parameters for ipropyl and tbutyl are in good agreement with the experimental values.
Keywords
O-alkyl S-methyl Xanthate; Pyrolysis; Dithiocarbonates; Elimination pathways; DFT Calculation
Introduction
The Chugaev elimination is broadly defined as the thermal decomposition of pyrolysis of a xanthate ester of an alcohol that contains at least hydrogen to produce an Olefin, Carbonyl sulphide and a thiol (Figure 1). This reaction is similar to the thermal decomposition of acetates, carbamates and carbonates to produces olefins.
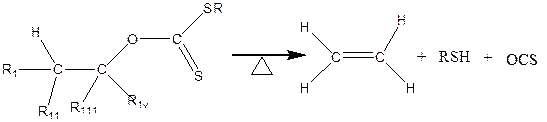
Figure 1. Chugaev Reactions [1] (where R 1, R11, R111 and Riv are the alkyl group under consideration)
The pyrolysis of carbonate related compounds (xanthates) were valuable synthetic routes to synthesis alkenes without the rearrangement of carbon skeleton [1-11]. Their mechanisms were long debated between two possible pathways (Figure 2) [7, 8], although both pathways resulted in the same products. The pathway reported in this research proceeded through a one-step mechanism yielding an alkene, carbonyl sulphide and thiol.
Cross et al., [12] and Huckel et al., [13] supported the pathway reported, while Smith and Yates [5], Barton [9], and Cram [6] preferred another mechanism. Experimentally, Al-Awadi and co-worker [14] pyrolyzed a series of the esters: thiocarbonates carbamates and carbonates. The pathway of six cyclic transition state approved by the positive p-factor of Hammett correction for the pyrolysis of aryl ethyl carbonates, as well as t-butyl, N-aryl carbonates with the reduced relativities of methyl alkyl carbonates as compared with phenyl alkyl carbonates [10, 11, 14-16]. Alexander and Mudrack also proved the other pathway, as well as the cis-elimination mechanism, by investigating the Chugaev reaction [17-19].
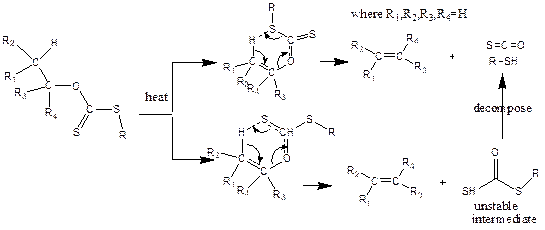
Figure 2. Mechanism of alkyl Xanthates
Bader and Borns [20] carried out isotope effect studies and found solid evidence for the pathway of six cyclic transition state. As for the theoretical studies, Lee group studied the pyrolysis of carbonate esters at AM1 level and supported the second pathway [21]. Erickson and Kahn [22] calculated the activation energies of HSC(=S) OEt at MP2/6-31G(d)/ HF/6-31g(d) level in 1994. Deleuze group [23] investigated the elimination reaction (Ei) reaction of EtSc(=S)OMe and its substituent effects at different theoretical levels and different basis sets and concluded that MP2 method gave comparable activation energies as CCSD(T). Harano [24] utilized B3LYP/6-31G(d) theoretical level to study the transition states of MeSc(=S)OEt. The regioselective pyrolysis of chugaev reaction was also investigated recently at the MP2/6-31G(d) level [25]. Consequently, this work aimed at studying the α- and β-elimination pathways (elimination of hydrogen attached to the α- carbon of the alkyl xanthate and β-carbon of the alkyl xanthate) and the effect of these elimination pathways on the kinetics and thermodynamic parameters using Spartan software with density functional theory method of calculations with Becke Lee Yang Parr number three at 6-311++G** level.
Computational Procedure
All calculations on the mechanisms, kinetics and thermodynamics of the gas- phase pyrolysis of α- and β- elimination pathways have been examined using the density functional theory (DFT) with B3LYP at the levels of 6-311++G** levels. Conformational search was done using molecular mechanics force field (MMFF) to obtain the most stable conformer [26, 27]. Geometry optimization was performed on the most stable conformer at the ground state, transition state and product to obtain the bond length, bond angle, dihedral angles and atomics charges distribution. Reaction path calculations were performed on the optimized geometry of each of α- and β-elimination path ways of the studied alkyl Xanthates using Spartan software [28]. Since these alkyl Xanthates followed the same pattern, O-ethyl S-methyl Xanthates was used as example, using H-S as the reaction coordinate. The internal coordinate varied systematically in many small steps from its initial distance in the stable reactants form to its stable product form for example, for O-ethyl, S-methyl xanthate, the initial distance between H and S is 4.8Å. This inter atomic distance is slowly altered throughout the reaction path calculation taking the value from 4.8 (initial value) to 1.2 Å its approximate value in the stable product molecule in 20 steps. Location and characterization of the transition state was done by subjecting the transition state structure to test in order to verify that this connect to true transition state and that this connect to the reactant and products. The test are: that there will be an imaginary IR value that will be in the range of 400-2000cm-1 and the normal coordinate must correspond to the imaginary frequency which will connect the reactants and the product. The intrinsic reaction coordinate method was also used. kinetics and thermodynamics calculations were done using the data obtained from the geometric calculations on the reactants, transition state and products. The kinetic and thermodynamics parameters were obtained using the sum of the ground state (GSE) and the statistical mechanically calculated enthalpy to arrive at a closer approximation of the true energy of the molecule.
Theoretical calculations
The enthalpy of a species was defined as:
![]() (1)
(1)
where the superscript sm is the statistical mechanically calculated enthalpy.
When Eq(1) was substituted into the initial definition of the heat of reaction we have,
![]() (2)
(2)
Activation energy (Ea) was calculated according to the transition state theory for a unimolecular reaction at 629K [27]:
Ea = ΔH* - RT (3)
The entropy of the reaction was calculated by taking the difference of product and reactant entropies that is:
ΔSreaction = Sproduct - Sreactant (4)
ΔSactivared = Stransition - Sreactant (5)
The Gibbs free energy (∆G) and Gibbs free energy of activation (∆G*) were calculated using the modified version of the heat of reaction equation, knowing that the first order coefficient K(T) was calculated using transition state theory (TST) [29] as shown below:
G = H - TS (6)
and
∆G* = ∆H* - T∆S (7)
where ∆G* is the Gibbs free energy change between the reactant and the transition state.
The Transition State Theory (TST) [10] was used to estimate the first-order rate coefficients k(T) and it was calculated assuming that the transmission coefficient is equal to 1, in the expression:
Pre-exponential factor is given as:
![]() (8)
(8)
where KB and h are the Boltzman and Planck constants respectively.
Arrhenius rate was obtained using the rate equation [29].
![]() (9)
(9)
The bond lengthening character
was used to describe the extent of bond lengthening to show the rate
determining step and to explain the concertedness and
synchronicity of the reaction. Moyano et al.
[30] have defined a relative variation of the bond indices at the reactant,
transition state and the product (![]() Bi)
for every ith bond involved in a chemical
reaction as:
Bi)
for every ith bond involved in a chemical
reaction as:
![]() (10)
(10)
where the superscripts R, TS and P refer to reactants, transition states and products respectively.
Also (%Ev) the percentage of evolution of the bond order during the calculated mechanism can be obtained from
![]() (11)
(11)
The dissociation of the Cα-O bond before Cβ-H bond is in accordance with the experimental proposal by Taylor et al. [16], for the 1, 5 thermal elimination based on the fact that the Hammelt P-values at the αcarbon were of larger magnitude than those observed for the βcarbon. The results also implies that, the more the Cα-O bond dissociate in the transition structure, the faster the rate of reaction and that the more the Cβ-H bond dissociate , the slower is the reaction.
The average value of the bond indices δBav is calculated as:
![]() (12)
(12)
where n in the above equation represented the number of bonds involved in the reaction coordinates.
The synchronicity (Sy), of a chemical reaction can be calculated as follows:
Sy = 1 A (13)
where A represents the asynchronicity. It is calculated by using the equation proposed by Moyano et al. [30]:
![]() (14)
(14)
Synchronicities vary between zero and one. This range indicate situation when all of the bonds at the reaction coordinate have been broken or formed at exactly the same extent in the transition states.
Results and discussion
Conformational search
Conformational Search was done using molecular mechanics force field (MMFF) showed that alkyl Xanthates under studied, have different conformers with the most stable conformer having energy given below in Table 1 and (Figure 3).
Table 1. Table of Number of conformers and stable conformers
with their corresponding Energy values of O-alkyl S-Methyl Xanthate
|
COMPOUND |
NO. OF CONFORMERS |
MOST STABLE CONFORMERS (kJ/mol) |
|
O-ethyl S-methyl Xanthate |
5 |
-48.389 |
|
O-npropyl S-methyl Xanthate |
12 |
-34.335 |
|
O-ipropyl S-methyl Xanthate |
7 |
-34.335 |
|
O-nbutyl S-methyl Xanthate |
31 |
-50.926 |
|
O-isobutyl S-methyl Xanthate |
10 |
-31.190 |
|
O-tbutyl S-methyl Xanthate |
4 |
16.061 |
|
a) o-ethyl s-methyl xanthate |
b) o-npropyl s-methyl xanthate |
|
c) o-nbutyl s-methyl xanthate |
d) o-ipropyl s-methyl xanthate |
|
e) o-ibutyl s-methyl xanthate |
f) o-tbutyl s-methyl xanthate |
Figure 3. Structure of most stable conformers of the alkyl Xanthate
Geometry optimization
Geometry Optimization was performed on each structure of alkyl Xanthate (Figure 4) in the ground state, transition state and the product to obtain the bond length, bond angles, dihedral angles and atomic charges distribution for the α- and β- elimination series. Changes were observed in bond lengths of the ground state, transition state and the product for of α - and β- elimination series as shown in table 2. The decrease in C-C bond length in transition state(1.406) compared to that of the ground state (1.517) is as a result of the release of electron from C-O and C-H (point of cleavages) in both α- and β- elimination series thus making the carbon-carbon (C-C) single bond at the ground state attained a carbon-carbon (C=C) double bond character at the transition state for α- and β- elimination series respectively. As shown in Figure 4 and Table 2, for α-elimination series using O-ipropyl and npropyl S-methyl xanthates in the TS, the long stretch in the bond lengths between C-H (from 1.094Å to1.248Å) and C-O (from 1.332Å to 2.291Å) are indications that cleavages occurs between C-H and C-O. Also the decrease in the inter-atomic distance of S-H from GS= 4.737Å to TS= 1.952Å and finally to Product = 1.348Å showed that a new bond is being formed.
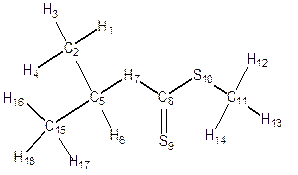
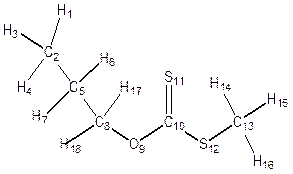
Figure 4. Geometry optimization of α- and β- O-isopropyl and O-npropyl S-methyl Xanthate
In the β- elimination series, the long stretch between C-H (from 1.0940Å to 1.2480Å and C-O (from 1.3319Å to 2.2910Å) it shows that cleavages also occurred at these point and the change in the change in bond length between S-H from GS= 4.7370Å bond, TS=1.9520Å and product= 1.3482Å are indications that a new bond has been formed.
Table 3 also shows the bond angles for the ground state, transition state and product, the variation in the values from ground state and transition state is as a result of distortion that occurred in structures during the transition process and this kind of distortion are necessary for intramolecular transfer of protons.
Table 2. Selected bond
length (Å) for the pyrolysis of o-alkyl s-methyl xanthates using
B3LYP/6-311++G**
|
Bond (Å) |
State |
Ethyl Xanthate |
nPropyl Xanthate |
ibutyl Xanthate |
iPropyl Xanthate |
ibutyl Xanthate |
|
H C |
GS TS PRD ∆d |
1.0940 1.2480 - 0.1540 |
1.0916 1.2350 - 0.1434 |
1.0927 1.2370 - 0.1443 |
1.0831 1.2470 - 0.1639 |
1.0832 1.2370 - 0.1538 |
|
C C |
GS TS PRD ∆d |
1.5170 1.4058 1.3308 - 0.1112 |
1.5143 1.4066 1.3306 - 0.1077 |
1.5134 1.4023 1.3306 - 0.1111 |
1.5133 1.3896 1.3171 - 0.1237 |
1.5136 1.3915 1.3182 - 0.1221 |
|
O C |
GS TS PRD ∆d |
1.3364 1.2471 - -0.089 |
1.3357 1.2473 - -0.0884 |
1.3362 1.2449 - -0.0913 |
1.3068 1.2163 - -0.090 |
1.3162 1.2146 - -0.1016 |
|
C = S |
GS TS PRD ∆d |
1.6473 1.7333 1.8142 0.086 |
1.6435 1.7269 1.8141 0.0834 |
1.6452 1.7220 1.7922 0.0768 |
1.6385 1.7311 1.7850 0.0926 |
1.6486 1.7264 1.7345 0.0778 |
|
S C |
GS TS PRD ∆d |
1.8189 1.8334 1.8284 0.0145 |
1.8165 1.8288 1.8288 0.0123 |
1.8270 1.8362 1.8361 0.0092 |
1.8053 1.8105 1.8111 0.0052 |
1.8064 1.8108 1.8107 0.0044 |
|
H S |
GS TS PRD ∆d |
4.9940 1.8160 1.3480 - 3.178 |
4.9950 1.8400 1.3481 - 3.155 |
4.9962 1.8260 1.3390 - 3.1702 |
4.910 1.8770 1.3250 - 3.033 |
4.9220 1.8980 1.3242 - 3.024 |
Reaction path study
Reaction path study were performed on the optimized geometry of each of the O-alkyl S-methyl Xanthate under study using α- and β-HS from O-ethyl S-Methyl dithiocarbonates as the reaction coordinate [26] the internal coordinate was varied from its initial distance in the stable reactants form to its value in the product molecule. As reported in literature [31], instead of the energy to pass smoothly through a maximum rose to a very high valve and the geometry suddenly drop to a product with a drop in the heat of formation with the values approximately the same as the sum of the expected produced (ethylene and methyl dithiocarbonates) as shown in Figures 58. The result obtained showed that the pyrolysis of O-alkyl S-methyl Xanthate can be initiated from the hydrogen attached to α-carbon of the O-alkyl moiety to give carbonyl sulphide thiol and olefin. It can also be initiated from the hydrogen attached to the β-carbon of the O-alkyl moiety to give carbonylsulphide, thiol and olefin.
Table 3. Selected bond
angle (0º) for the pyrolysis of o-alkyl s-methyl xanthates using
B3LYP/6-311+G**
|
Bond angle (0º) |
State |
Ethyl Xanthate |
nPropyl Xanthate |
ibutyl Xanthate |
iPropyl Xanthate |
ibutyl Xanthate |
|
H C C |
GS TS PRD |
110.70 102.74 - |
110.79 102.74 - |
110.820 102.720 - |
110. 573 98.09 - |
110.567 97.240 - |
|
C C O |
GS TS PRD |
106.95 108.95 - |
106.97 109.18 - |
107.820 109.320 - |
106.655 108.910 - |
106.620 108.930 - |
|
C O C |
GS TS PRD |
122.82 118.90 - |
122.88 120.32 - |
122.745 119.090 - |
124.951 123.810 - |
124.920 122.930 - |
|
O - C = S |
GS TS PRD |
120.00 124.78 124.51 |
120.11 125.16 123.61 |
120.151 124.914 124.620 |
120.051 124.734 123.960 |
120.120 124.311 124.970 |
|
O - C S |
GS TS PRD |
114.57 120.47 123.97 |
114.48 119.14 120.06 |
120.575 120.575 122.720 |
114.894 119.351 120.060 |
114.821 119.621 121.240 |
|
S C S |
GS TS PRD |
125.45 114.73 111.52 |
125.40 115.70 112.63 |
114.488 114.488 114.72 |
125.054 115.914 116.290 |
12.050 116.062 116.520 |
|
C S C |
GS TS PRD |
101.96 102.15 98.140 |
102.30 101.41 98.120 |
102.321 102.938 97.620 |
103.678 101.039 99.330 |
103.620 101.530 99.440 |
|
S C H |
GS TS PRD |
110.39 109.48 110.28 |
110.36 110.98 111.24 |
110.470 109.333 112.462 |
110.708 110.771 110.360 |
110.765 110.740 111.360 |
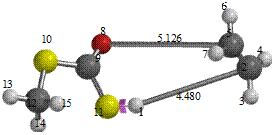
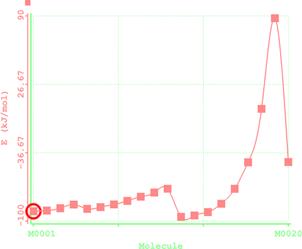
Figure 5. Reaction path study (O-ethyl S-methyl Dithiocarbonates) and a plot of energy against number of molecules to depict the reaction path study
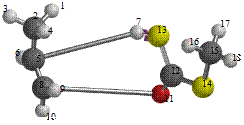
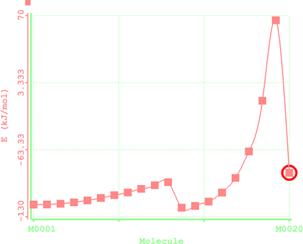
Figure 6. Reaction path study O-npropyl S-methyl Dithiocarbonates) and a plot of energy against number of molecules to depict the reaction path study
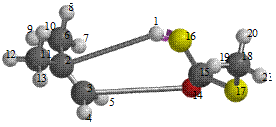
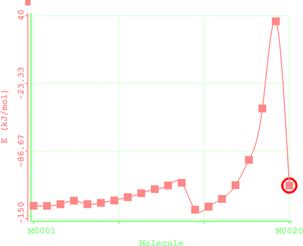
Figure 7. Reaction path study O-ipropyl S-methyl Dithiocarbonates) and a plot of energy against number of molecules to depict the reaction path study
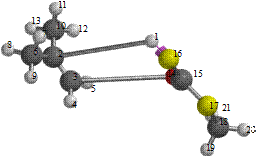
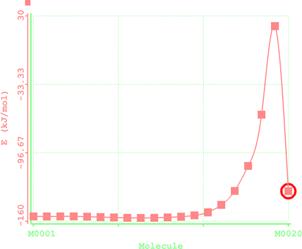
Figure 8. Reaction path study O-ibutyl S-methyl Dithiocarbonates) and a plot of energy against number of molecules to depict the reaction path study
Transition state
The transition state was characterized using two methods, the infrared (IR) values obtained for α- and β-elimination which has only one imaginary IR frequency values. The main components of these stretching are SH. The CO stretching is not present because the bond is virtually broken. The intrinsic reaction coordinate calculation converged, which shows that reaction is an exothermic one.
Mechanism of pyrolysis of O-alkyl S-methyl xanthate
The pyrolysis of O-alkyl S-methyl Dithiocarbonates (Xanthate) has been reported to proceeds through a concerted sixcentered cyclic transition state initiated from the hydrogen that is attached to either the α- and β-carbon of the O-alkyl xanthate in which O-ethyl S- methyl xanthate is the simplest of the α- and β- studied which gives Methyl dithiocarbonates and ethylene (C2H4). However the result obtained for other pyrolysis of α-elimination series are O-isopropyl xanthate and O-tbutyl xanthate and the elimination series are O-npropyl xanthate, O-ibutyl xanthate and O-nbutyl xanthate are given below in Table 4.
Table 4. Reaction Mechanisms of O-alkyl Xanthate showing the
α- and β-carbon elimination series
|
α-Elimination series |
β -Elimination series |
||
|
Reactant |
Products |
Reactant |
Products |
|
O-isopropyl Xanthate |
Propene, thiol and Carbonylsulphide |
O-npropyl Xanthate |
Propene, thiol and Carbonylsulphide |
|
O-tbutyl Xanthate |
2-methyl prop-1-ene, thiol carbonylsulphide |
O-isobutyl Xanthate |
2-methylprop-1-ene, thiol carbonylsulphide |
|
O-nbutyl Xanthate |
2-methylprop-1-ene, thiol carbonylsulphide |
||
There are various studies on the mechanism of the thermal decomposition of xanthate. The mechanism of the Chugaev reaction was proposed to involve two possible pathways [13]. The Chugaev reaction is analogous to the thermal decomposition of carboxylic ester of alcohols and of other related derivatives of alcohols such as carbamates and carbonates. The mechanism of xanthate pyrolysis is a concerted fragmentation but products could conceivably arise from α- and β-hydrogen abstraction through a six-membered Cyclic transition State in which the βH atom attacking the terminal thiol, or the thion [13]. Sulphur atom attack the βhydrogen which was obtained by Bader and Bourns [20], both the thiol and thion were part of the six membered cyclic transition State which involves an SH bond making, CH and CO bond breaking and according to experimental evidence, the C-O bond breaking is the rate determining step [32].
Atomic charges
The charges (Table 5) showed that α-H and β-H has positive charge development (+0.191 and +0.155) while S has negative charges (-0.492 and -0.550) using isopropyl and npropyl xanthate (Figure 9). The polarization of C-O and S-H bonds causes positive charges on carbon-carbon bonds (+0.138 and +0.410) respectively and causes C to become negatively charged (-0.127) for α-elimination series and the polarization of the S-H and C-O causes positive charges on carbon-carbon to increase respectively.
Table
5. Atomic charges (Mulliken) of o-alkyl s-methyl
xanthates using MP2/B3LYP/6-311+G**
|
Atoms |
State |
Ethyl Xanthate |
nPropyl Xanthate |
ibutyl Xanthate |
iPropyl Xanthate |
tbutyl Xanthate |
|
H |
GS TS PRD |
+0.174 +0.141 +0.273 |
+0.155 +0.176 +0.210 |
+0.191 +0.116 +0.221 |
+0.191 +0.238 +0.096 |
+0.193 +0.195 +0.095 |
|
∆q(TS-GS) |
-0.033 |
0.021 |
-0.075 |
0.047 |
0.002 |
|
|
C |
GS TS PRD |
-0.458 -0.480 -0.285 |
-0.550 -0.351 -0.295 |
-0.560 -0.285 -0.321 |
-0.492 -0.619 -0.353 |
-0.462 -0.391 -0.299 |
|
∆q(TS-GS) |
-0.022 |
-0.199 |
0.275 |
-0.127 |
0.071 |
|
|
C |
GS TS PRD |
-0.071 -0.123 -0.285 |
-0.256 -0.198 -0.296 |
-0.242 -0.047 -0.286 |
+0.043 -0.052 -0.353 |
+0.044 -0.062 -0.362 |
|
∆q(TS-GS) |
-0.052 |
0.058 |
0.195 |
0.095 |
-0.106 |
|
|
O` |
GS TS PRD |
-0.387 -0.437 -0.389 |
+0.120 -0.126 -0.426 |
+0.140 -0.330 -0.520 |
-0.537 -0.614 -0.362 |
-0.620 -0.243 -0.426 |
|
∆q(TS-GS) |
-0.050 |
-0.246 |
-0.470 |
-0.077 |
0.377 |
|
|
C |
GS TS PRD |
+0.064 +0.082 +0.098 |
-0.009 -0.011 +0.068 |
-0.016 +0.156 +0.727 |
-0.151 +0.190 +0.177 |
+0.162 +0.171 +0.182 |
|
∆q(TS-GS) |
0.018 |
0.002 |
0.172 |
0.341 |
0.009 |
|
|
S |
GS TS PRD |
+0.162 +0.164 +0.158 |
-0.094 +0.061 +0.168 |
-0.093 +0.002 +0.268 |
+0.192 +0.171 +0.041 |
+0.182 -0.017 +0.042 |
|
∆q(TS-GS) |
-0.021 |
-0.155 |
0.095 |
-0.021 |
-0.199 |
|
|
S |
GS TS PRD |
-0.186 -0.116 -0.011 |
-0.310 -0.244 -0.012 |
-0.326 -0.256 -0.014 |
-0.230 -0.263 +0.152 |
-0.241 -0.417 +0.156 |
|
∆q(TS-GS) |
0.07 |
0.066 |
0.070 |
-0.033 |
-0.176 |
|
β-elimination series (O-npropyl xanthate) |
α-elimination series (O-npropyl xanthate) |
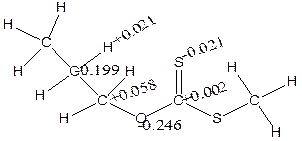
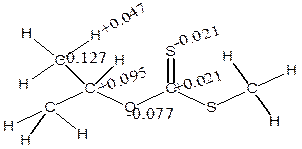
Figure 9. The distribution of atomic charges for α- and β- elimination series
Kinetics and thermodynamics parameters calculation
The result obtained for the kinetics and thermodynamics parameters (Table 6) for the β elimination pathways for ethyl, npropyl, ibutyl and nbutyl respectively ranged from 161.34-128.04kJ/mol(∆H*), -31.18 to -24.00 J/mol/K(∆S*), 180.65 to 142.92 kJ/mol(∆G*), 166.49-133.19 kJ/mol(Ea), 1.04x10-3 to 4.3 S-1 (K620) and 3.06x 1011to 7.18 x 1011(A). It was observed that the progressive methylation decreases activation and thermodynamics parameters from ethyl, propyl, isopropyl, nbutyl, ibutyl and tbutyl xanthate respectively. However, progressive methylation increases the rate constant (k), and pre-exponential factor (A) respectively. Also for α elimination pathway kinetic and thermodynamic parameters for ipropyl and tbutyl compared favourably well with the experimental values.
Table 6. Effect of methylation on kinetic and
thermodynamic parameters for α and βelimination
series at 629K (B3LYP/6-311++G**)
|
CALCULATED PARAMETERS |
||||||
|
Compounds |
∆H* kJ/mol |
∆S* J/mol |
∆G* kJ/mol |
Ea kJ/mol |
A×1011 |
k620 (S-1) |
|
O-Ethyl Xanthate |
161.34 |
-31.16 |
180.65 |
166.49 |
3.06 |
1.04·10-3 |
|
O-nPropyl Xanthate |
153.23 |
-27.18 |
170.08 |
158.38 |
4.90 |
6.0·10-2 |
|
O-iPropyl Xanthate |
144.21 |
-26.98 |
160.94 |
149.36 |
5.00 |
1.3·10-1 |
|
O-nButyl Xanthate |
133.87 |
-25.65 |
149.77 |
139.02 |
5.89 |
1.14 |
|
O-iButyl Xanthate |
131.67 |
-25.78 |
147.65 |
136.82 |
5.81 |
1.70 |
|
O-tButyl Xanthate |
128.04 |
-24.00 |
142.92 |
133.19 |
7.18 |
4.3 |
|
EXPERIMENTAL PARAMETERS |
||||||
|
|
∆H kJ/mol |
∆S* J/mol |
∆G* kJ/mol |
Ea kJ/mol |
A×1011 |
k620 (S-1) |
|
O-Ethyl Xanthate |
161.00 |
-28.00 |
178.6 |
166.20 |
4.7 |
1.4·10-3 |
|
O-nPropyl Xanthate |
|
|
|
|
|
|
|
O-iPropyl Xanthate |
- |
- |
- |
- |
- |
- |
|
O-nButyl Xanthate |
144.00 |
-26.00 |
160.4 |
149.17 |
5.7 |
5.6·10-1 |
|
O-iButyl Xanthate |
- |
- |
- |
- |
- |
|
|
O-tButyl Xanthate |
- |
- |
- |
- |
- |
- |
It was further observed that addition substituents to the βcarbon lower the activation energy slightly from ethyl xanthate to i-butyl xanthate) with a difference of about 29.67kJ/mol., while the addition of substitutents to the αcarbon from ethyl xanthate to t-butyl xanthate lower the activation energy the more, with a difference of about 33.45kJ/mol.
Wiberg bond order analysis for o-alkyl s-methyl dithiocarbonates
Generally, the calculated percentage of evolution of the bonds involved in the reaction coordinates is collected in Table 7.
Table 7. Calculation
using Wiberg bond indices Bi of reactants,
transition and products, percentage of evolution of bonds %Ev,
degree of advancement of the transition states, δBav
and absolute synchronicities Sy, of O-alkyl
S-methyl Xanthate (B3LYP/6-311++G**)
|
Compound |
Wiberg bond indices |
S1-C2 |
C2-O3 |
O3-C4 |
C4-C5 |
C5-H6 |
H6-S1 |
|
O-Ethyl Xanthate |
BiR |
1.678 |
1.034 |
0.813 |
1.030 |
0.935 |
0.000 |
|
BiTS |
1.332 |
1.390 |
0.279 |
1.341 |
0.534 |
0.347 |
|
|
BiP |
1.070 |
1.695 |
0.000 |
2.034 |
0.000 |
0.962 |
|
|
δBi |
0.57 |
0.539 |
0.658 |
0.310 |
0.429 |
0.360 |
|
|
%Ev |
57 |
53.9 |
65.8 |
31 |
42.9 |
36.0 |
|
|
O-nPropyl Xanthate |
BiR |
1.018 |
0.928 |
0.002 |
1.666 |
1.041 |
0.790 |
|
BiTS |
1.310 |
0.549 |
0.325 |
1.319 |
1.407 |
0.225 |
|
|
BiP |
1.985 |
0.000 |
0.962 |
1.070 |
1.695 |
0.000 |
|
|
δBi |
0.317 |
0.408 |
0.716 |
0.582 |
0.560 |
0.317 |
|
|
%Ev |
30.2 |
58.2 |
71.6 |
58.2 |
40.9 |
31.7 |
|
|
O-iPropyl Xanthate |
BiR |
1.666 |
0.926 |
0.771 |
1.007 |
0.005 |
0.921 |
|
BiTS |
1.321 |
0.435 |
0.179 |
1.274 |
0.298 |
0.570 |
|
|
BiP |
1.070 |
0.000 |
0.000 |
1.934 |
0.962 |
0.000 |
|
|
δBi |
0.579 |
0.530 |
0.768 |
0.288 |
0.310 |
0.390 |
|
|
%Ev |
57.9 |
53.0 |
76.8 |
28.8 |
31.0 |
39.0 |
|
|
O-nButyl Xanthate |
BiR |
0.980 |
0.926 |
0.771 |
0.980 |
0.005 |
1.016 |
|
BiTS |
0.309 |
0.435 |
0.357 |
0.309 |
0.298 |
1.411 |
|
|
BiP |
0.000 |
0.000 |
0.000 |
0.000 |
0.962 |
1.983 |
|
|
δBi |
0.685 |
0.530 |
0.76 |
0.685 |
0.310 |
0.407 |
|
|
%Ev |
68.5 |
53.0 |
76.0 |
68.5 |
31.0 |
40.7 |
|
|
O-iButyl Xanthate |
BiR |
1.666 |
1.045 |
0.771 |
1.006 |
0.006 |
0.003 |
|
BiTS |
1.321 |
1.419 |
0.179 |
1.273 |
0.299 |
0.492 |
|
|
BiP |
1.070 |
1.695 |
0.000 |
1.933 |
0.964 |
0.953 |
|
|
δBi |
0.579 |
0.576 |
0.769 |
0.287 |
0.320 |
0.361 |
|
|
%Ev |
57.9 |
57.6 |
77.8 |
28.7 |
32.0 |
51.50 |
|
|
O-tButyl Xanthate |
BiR |
1.666 |
1.045 |
0.771 |
1.007 |
0.921 |
0.005 |
|
BiTS |
1.322 |
1.419 |
0.179 |
1.274 |
0.570 |
0.298 |
|
|
BiP |
1.070 |
1.695 |
0.000 |
1.934 |
0.000 |
0.962 |
|
|
δBi |
0.579 |
0.576 |
0.768 |
0.288 |
0.390 |
0.31 |
|
|
%Ev |
57.9 |
57.6 |
76.8 |
28.8 |
39.0 |
31.0 |
As it can be seen, in the case of O-alkyl S-methyl Dithiocarbonates, (1-6), in which alkyl group are ethyl, npropyl, isopropyl, nbutyl, ibutyl and tbutyl, the breaking of the bond O-C is the most advanced process (65-77)%, followed by the transformation of the S-C double bond into a single bond (58%) and of the C-O single bond into double bond of (54-58)%.
The least advanced process is the formation of the C-C double bonds (30%) followed by the formation of the H-S bond (31-36)% and the breaking of the C-H bond (39-43)%. The dissociation of the Cα-O bond before Cβ-H bond is in accordance with the experimental proposed by Taylor and co-worker in 1962 [16], for the 1, 5 thermal elimination. The results also implies that the more the Cα-O bond is dissociated in the transition structure the faster is the rate of reaction and that the more the Cβ-H bond is dissociated , the slower is the reaction. This trend in bond lengthening can be mirrored in the atomic charges. In the table of atomic charges at the atoms involved in the reaction centers of mechanism for all the compound studies.
There is a buildup of negative charge on Oxygen atom in each transition structure, consistent with C-O bond dissociation that increase rate, in accordance with the above observation that the C-O bond dissociated to a larger degree in the faster reactions. The opposite trend is apparent in the buildup of positive charge on the transferred hydrogen that decreases as the rate increases, in agreement with the trends in CH bond lengthening [22]. The calculated δBav values for the first step of the mechanism of the studied reactions are shown in table 8. As it can be seen, the δBav values show that there is a difference between the transition states of the first mechanism (B) of the Chugaev reaction for the Xanthate considered. In the case of the O-alkyl S-methyl dithiocarbonates Bav values ranges from 0.478 to 0.485, increasing from O-ethyl to O-tert-butyl, indicating that the transition states have an early character, nearer to the reactants than to the products.
Synchronicities vary between zero and one, which is the case when all of the bonds at the reaction coordinate have been broken or formed at exactly the same extent in the transition states. The Sy value obtained for O-alkyl S-methyl dithiocarbonates range from 0.807 to 0.860 (Table 8) for the reaction of the compounds, indicating that O-alkyl S-methyl dithiocarbonates corresponds to highly asynchronous processes. The synchronicity rapidly decreases when the size of the alkyl group attached to sulphur or oxygen atoms increases.
Table
8. Calculation using Wiberg, degree of
advancement of the transition states, δBav
and absolute synchronicities Sy, of
O-alkyl S-methyl Xanthate (B3LYP/6-311++G**)
|
Compound |
δBav |
Sy |
|
O-Ethyl Xanthate |
0.478 |
0.860 |
|
O-nPropyl Xanthate |
0.496 |
0.832 |
|
O-iPropyl Xanthate |
0.484 |
0.807 |
|
O-nButyl Xanthate |
0.486 |
0.867 |
|
O-iButyl Xanthate |
0.485 |
0.807 |
|
O-tButyl Xanthate |
0.485 |
0.807 |
Conclusions
The calculations related with the reaction path showed that the gas-phase pyrolysis could conveniently proceeds through the two pathways that is elimination initiated from the hydrogen that is attached to α-carbon of the O-alkyl xanthates and from the hydrogen that is attached to the β-carbon of the O-alkyl xanthates. It is observed that ab-initio method could effectively predict the kinetics, mechanisms and thermodynamics of the gas-phase pyrolysis of an alkyl Xanthate. In addition, the computed Arrhenius and thermodynamic parameters obtained at 629K kelvin are in good agreement with experimental values. The calculated percentages of evolution of the bonds involved in the reaction indicate that the bond breaking for these compounds and the bond breaking for these compounds are the most advanced processes for the reactions in the transition states. However, the energy of formation and other calculated kinetic and thermodynamic parameters for the elimination pathway from the hydrogen that is attached to α-carbon of the O- alkyl xanthates is lower compared with that of the β-carbon of the O-alkyl xanthates. Useful kinetic and thermodynamics parameters can be obtained theoretically prior to their laboratory experimentations.
References
1. Tschugaeff L., Chugaev, Bericcte Der Deutschen Chemistchen Gesellschaft, 1899, 32, p. 3332-3335.
2. Tschugaeff L., Chugaev, Bericcte Der Deutschen Chemistchen Gesellschaft, 1898 p. 17751782.
3. Smith G. G., Jones D. A. K., Taylor R., Pyrolysis studies. IX. Rates and mechanism of thermal decomposition of phenyl and benzyl ethyl carbonates, Journal of Organic Chemistry, 1963, 28, p. 3547-3550.
4. Smith G. G., Lum K. K., Kirby J. A., Posposil J., Linear free energy relation involving ortho substituents in gas phase reactions. XVII, Journal of Organic Chemistry, 1969, 34, p. 2090-2095.
5. Smith G. G., Yates B. L., Kinetics of vapor phase pyrolysis of aryl ethyl methyl carbonates. A linear free-energy relationship for ortho substituents, Journal of Organic Chemistry, 1965, 30 p. 434-438.
6. Cram D. J., Studies in stereochemistry. IV. The Chugaev reaction in the determination of configuration of certain alcohols, Journal of the American Chemical Society, 1947, 71, p. 3883-3889.
7. OConnor G. L., Nace H. R., Further studies on the Chugaev reaction and related reactions, Journal of the American Chemical Society, 1953, 75, p. 2118-2123.
8. Tsou K.-C., Seligman A. M., Preparation of alkyl 2-naphthyl carbonates as chromogenic substrates for Esterases1, Journal of the American Chemical Society, 1954, 76, 3704-3706.
9. Barton D.H.R., cis-Elimination in thermal decompositions, Journal of Chemical Society 1949, 464, p. 2174-2178.
10. Alawadi N., Bigley D. B., Gas-phase pyrolysis of some unsymmetrical monothiolcarbonates and a rationalization of the rates of some related reactions, Journal of Chemical Society, Perkin Transaction, 1979, 2, p. 497-500.
11. Alawadi N., Bigley D. B., Carbonate pyrolysis 6. The kinetics and mechanism of the pyrolysis of thio-carbonate and dithio-carbonate - implications for the transition-state in carbonate pyrolysis, Journal of Chemical Society, Perkin Transaction, 1982, 2, p. 773-775.
12. Cross J. T. D., Hunter R., Stimson V. R., Thermal-decomposition of simple carbonate esters, Australian Journal of Chemistry, 1976, 29, p. 1473-1481.
13. Hückel W., Tappe W., Legutke G., Abspaltungsreaktionen und ihr sterischer Verlauf, Annalen der Chemie, 1940, 543, p. 191-230.
14. Alawadi N., Taylor R., The mechanism of thermal eliminations; rate data for pyrolysis of S-aryl O-ethyl thiocarbonates - the relative abilities of oxygen and sulfur to transmit electronic effects, Journal of Chemical Society, Perkin Transaction 2, 1986, p. 1581-1583.
15. Alawadi N., Taylor R., The mechanism of thermal eliminations, Elimination from monothiocarbonates, dithiocarbonates, and trithiocarbonates - the dependence of the transition state polarity, thione to thiol rearrangement, and ether formation via nucleophilic-substitution, on compound type, Journal of Chemical Society, Perkin Transaction 2, 1988, p. 177-182.
16. Taylor R., The mechanism of thermal eliminations, Journal of Chemical Society, Perkin Transaction 2, 1983, p. 291-296.
17. Alexander E. R., Mudrak A., Studies on the mechanism of Chugaev and acetate thermal decompositions. I. cis- and trans-2-Phenylcyclohexanol, Journal of the American Chemical society, 1950, 72, p. 1810-1813.
18. Alexander E. R., Mudrak A., Studies on the mechanism of Chugaev and acetate thermal decompositions. II. cis- and trans-2-Methyl-1-tetralol, Journal of the American Chemical society, 1950, 72, p. 3194-3198.
19. Alexander E. R., Mudrak A., Studies on the mechanism of Chugaev and acetate thermal decompositions. III. cis and trans-2-Methyl-1-indanol, Journal of the American Chemical society, 1951, 73, p. 59-62.
20. Bader R. F. W., Bourns A. N., A kinetic isotope effect study of the Tschugaeff reaction, Canadian Journal of Chemistry, 1961, 39, p. 348-358.
21. Lee I., Cha O. J., Lee B. S., Theoretical studies on the gas phase pyrolysis of esters, Journal of Physical Chemistry, 1990, 10, p. 3926-3930.
22. Erickson J. A., Kahn S. D., Theoretical studies of thermal syn elimination reactions. The relative rates of ethyl formate, ethyl xanthate, and ethyl Phosphinates eliminations, Journal of the American Chemical Society, 1994, 116, p. 6271-6276.
23. Claes L., François J. P., Deleuze M., Theoretical study of the internal elimination reactions of xanthate precursors, Journal of Computational Chemistry, 2003, 24, p. 2023-2031.
24. Harano K., Cascade reactions of unsaturated xanthates and related reactions: computer-assisted molecular design and analysis of reaction mechanisms, Yakugaku Zasshi, 2005, 125, p. 469-489.
25. Wu P., Truong J., Huang Y., Li J., Regioselectivity investigation for the pyrolysis of xanthates: a computational study, Journal of Theoretical and Computational Chemistry, 2013, 12, 1350064.
26. Adejoro I. A., Bankole T. O., Semi-Empirical Quantum Mechanical, Molecular orbital method using MOPAC, Calculation of the Arrhenius parameters for the pyrolysis of some alkyl Acetates, Journal of Applied Sciences, 2005, 5, p. 1559-1563.
27. Hehre W., Ohlinger S., Spartan06 for Windows, Machintosh and Linux tutorial and users guide. Wave function Inc. USA, 2006-2009.
28. Hehre W., Ohlinger S., Spartan10 for Windows, Machintosh and Linux tutorial and users guide. Wave function Inc. USA, 2010.
29. Benson S. W., ONeal, kinetic Data of Gas phase unimolecular reactions NSRDS-MBS 21-1970.
30. Moyano A., Pericas M. A., Valenti E., A theoretical study on the mechanism of the thermal and the acid-catalysed decarboxylation of 2-oxetanones (beta lactones), Journal of Organic Chemistry, 1989, 54, p. 573-582.
31. Wright M. R., An introduction to chemical Kinetics, John. Wiley & Sons England, 2004.
32. Mclever J. W., Kormonicki A. Rapid Geometry optimization for semi-empirical molecular orbital Models Chemical Physics Letters, 1971, 10, p. 303.